
Introduction
In this article, xenon’s medical effect is thoroughly studied. Xenon proves to have positive analgesic(pain relieving), organ protective and neuroprotective properties that make it potentially more medically useful and popular as an anesthetic and more.
Citation
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}0 Journal Article
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}A Preckel, Benedikt
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}A Weber, Nina C.
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}A Sanders, Robert D.
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}A Maze, Mervyn
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}A Schlack, Wolfgang
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}A Warltier, David C.
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}T Molecular Mechanisms Transducing the Anesthetic, Analgesic, and Organ-protective Actions of Xenon
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}B Anesthesiology
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}D 2006
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}R 10.1097/00000542-200607000-00029
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}J Anesthesiology
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}V 105
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}N 1
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}P 187-197
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}@ 0003-3022
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}X The anesthetic properties of xenon have been known for more than 50 yr, and the safety and efficacy of xenon inhalational anesthesia has been demonstrated in several recent clinical studies. In addition, xenon demonstrates many favorable pharmacodynamic and pharmacokinetic properties, which could be used in certain niche clinical settings such as cardiopulmonary bypass. This inert gas is capable of interacting with a variety of molecular targets, and some of them are also modulated in anesthesia-relevant brain regions. Besides these anesthetic and analgesic effects, xenon has been shown to exert substantial organoprotective properties, especially in the brain and the heart. Several experimental studies have demonstrated a reduction in cerebral and myocardial infarction after xenon application. Whether this translates to a clinical benefit must be determined because preservation of myocardial and cerebral function may outweigh the significant cost of xenon administration. Clinical trials to assess the impact of xenon in settings with a high probability of injury such as cardiopulmonary bypass and neonatal asphyxia should be designed and underpinned with investigation of the molecular targets that transduce these effects.
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}[ 6/26/2021
{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}U https://doi.org/10.1097/00000542-200607000-00029
Article

THE noble gas xenon has been known for more than 50 yr to have anesthetic properties1; however, its clinical utility has been limited by relatively high manufacturing costs owing to its rarity in the atmosphere.2Recently, the safety and efficacy of xenon inhalational anesthesia has been demonstrated in a variety of clinical settings3,4; in particular, xenon possesses favorable pharmacokinetic,4,5analgesic,6–8cardiovascular,3,9,10and safety properties.5,11Despite these desirable attributes, which make it an attractive anesthetic agent,5its high cost outweighs its routine use for general anesthesia.
Xenon is often referred to as inert because it is not transformed under biologic conditions; however, xenon is capable of interacting with a variety of molecular targets that may translate into desirable benefit for patients at risk of acute injury to the cardiovascular or nervous system or both.5,12In this review, we summarize the current data that have led us to believe that xenon could be used in the future to protect the heart and brain both in surgical and nonsurgical settings.
Physical and Chemical Properties
Xenon is the 54th element in the Periodic Table of the Elements and exists as a monoatomic gas. In common with the other inert gases, its outer shell is completely filled with electrons. Therefore, xenon has a low propensity to receive or release electrons, making it unlikely to form covalent bonds. Only under extreme nonbiologic conditions can halides of xenon (e.g. , XeF2, XeF4) be created. Xenon has a low ionization potential, allowing its electron shell to be polarized by surrounding molecules, thereby inducing a dipole that enables biologic interactions, including binding to proteins.13Xenon can associate with amino acid side chains of the active site of enzymes like serine proteinases (including elastases and collagenases)14,15; these enzymes can form a specific binding cavity for one single xenon atom without inducing major changes in protein structure.14It has been demonstrated that xenon binds within the heme cavity of cytochrome P-450 monooxygenases and is capable of inhibiting the catalytic activity of some enzymes in vitro .16
Putative Sites of Anesthetic Action
While several molecular targets in anesthesia-relevant brain regions are modulated by xenon, there is still no direct evidence to support one mechanism, although the role of glutamate receptors seems to be a pivotal one from recent studies involving Caenorhabditis elegans .17,18
Receptor Effects
Most general anesthetics act on one or more superfamilies of ligand-gated ion channels; e.g. , the γ-aminobutyric acid type A (GABAA), glycine, 5-hydroxytryptamine type 3A, and neuronal nicotinic acetylcholine (nACh) receptors are targets for several general anesthetics, including barbiturates, propofol, benzodiazepines, and halogenated inhalational agents.19Recent data has indicated that xenon induces anesthesia in a unique way by inhibiting excitatory glutamatergic signaling,18although it remains unclear which subtype of glutamate-gated receptors is responsible for xenon’s effects. This is certainly a feasible mechanism of anesthesia20,21and also has potential to explain the difference in potency between halogenated agents and xenon. Which of the three subtypes of postsynaptic glutamate-gated ion channels (referred to by its most selective ligand, namely N -methyl-d-aspartate [NMDA], kainic acid, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA] receptors) is the prime target is currently under debate. Recent data from an elegant series of experiments in C. elegans has shown that inhibition of non-NMDA receptors mediate the “anesthetic” effects of xenon. Using sophisticated pharmacogenomic techniques, Crowder et al. 18demonstrated that mutation of the glr-1 glutamate receptor subunit (homolog of the AMPA subunit Glur1) reduced xenon’s ability to induce “anesthesia.” Mutation of nmr-1 (which encodes the pore-forming subunit of the NMDA receptor in C. elegans ) did not affect the behavioral effects induced by xenon. As pointed out by the authors, multiple caveats must be introduced when interpreting these effects; e.g. , “anesthesia” in C. elegans is a change in behavioral phenotype that is not necessarily analogous to anesthesia in humans. Furthermore, there is a huge difference in genotype between humans and C. elegans . Nonetheless, the work supports the notion that xenon induces anesthesia by inhibiting glutamatergic signaling.
Xenon has been shown to noncompetitively block the NMDA subtype of the glutamate receptor in cultures of rat hippocampal neurons22; contrastingly, the fast component of the glutamate postsynaptic current that is mediated by the AMPA receptor was not affected.22,23Xenon did inhibit the current generated when the artificial agonist kainate is directly applied to recombinant AMPA receptors24; however, when the receptor was activated by its natural agonist glutamate using an ultrarapid application system to outside-out membrane patches to mimic synaptic conditions, the sensitivity of this subtype to xenon was negligible. Weigt et al. 25recently demonstrated that xenon inhibits AMPA- and kainate-induced membrane currents in cultured cortical neurons when glutamate was applied to whole cells using a slower application time. However, it seems likely that, under conditions that mimic natural synapses in mammalian systems,23,24non-NMDA receptors are insensitive to xenon. When NMDA receptors were expressed in Xenopus oocytes, xenon again inhibited NMDA receptor currents. The controversy will continue as to whether non-NMDA receptors are important targets for xenon; however, the current evidence firmly indicates that xenon inhibits NMDA receptor signaling, and this is regarded as the prime mechanism by which xenon induces anesthesia.
Xenon has little or no effect on the inhibitory GABAAreceptors in cultured rat hippocampal neurons,22which are quite sensitive to several other gaseous anesthetics.19There was no effect of xenon on GABAergic inhibitory postsynaptic currents or on currents evoked by exogenous application of GABA in cultured neurons containing excitatory and inhibitory synapses.23However, in recombinant GABA receptor complexes expressed in human embryonic kidney cells and Xenopus oocytes, xenon enhanced the inhibitory GABAergic transmission.26,27In human homomeric glycine receptors, xenon potentiated the current response to applied glycine, suggesting a contribution to the prolongation of the inhibitory postsynaptic potential.28However, because xenon exerts little effect on inhibitory neurotransmission in neuronal systems, there is currently little evidence to suggest that effects at GABA and glycine receptors contribute to the xenon anesthetic state.
The nACh receptors are found in presynaptic and postsynaptic locations within the central nervous system, acting by modulating transmitter release.29Various combinations of subunits of the nACh receptor are known, and it has been shown that isoflurane and propofol inhibited the most prevalent neuronal subtype (α4)2(β2)3of the nACh receptor but had no effect on the (α7)5nACh receptor subtype, even at high concentrations.30In contrast, halothane inhibited both receptor subunits.29Xenon inhibited (α4)2(β2)3nACh receptors expressed in Xenopus oocytes, whereas the α4β4nACh receptor was only slightly affected.27These data were extended by findings of Suzuki et al. ,31who demonstrated that xenon reversibly inhibited the ACh-induced currents in human homomeric (α7)5nACh receptors in a concentration-dependent manner. This effect was noncompetitive and voltage independent.31Despite the high sensitivity of nACh receptors to anesthetic agents, effects at this receptor are not thought to be critical for anesthesia.32Xenon at clinical relevant concentrations competitively inhibited the 5-hydroxytryptamine type 3A receptor independently of the membrane potential.33The clinical consequence of this effect is unknown.
The two-pore-domain potassium channel (so named because of two pore-forming consensus regions identified in their primary sequence) has been recently proposed as a target for general anesthesia.34–36Some members of this superfamily, the TREK-1 and the TASK-3, are activated by halogenated anesthetics like halothane.37Gruss et al. 34demonstrated that xenon is as effective as halothane in activating TREK channels. However, in contrast to the potent halogenated anesthetic halothane, the gaseous anesthetic had no effect on TASK channels. Similar to halogenated anesthetics,38xenon interacts with the cytoplasmic C-terminus of TREK channels, but this region is unlikely to contain primary binding sides for the atom.34The amino acid Glu306 has been found to play a major role in modulation of TREK-1 by arachidonic acid and membrane stretch and may be important for the activating effects of xenon on these channels.34
Second Messenger Signaling
General anesthesia may result from interference at the synaptic level of Ca2+-dependent transmitter release; in addition, modulation of second messenger systems may alter postsynaptic neuronal responses to released neurotransmitter. Changes in neuronal Ca2+homeostasis may alter neurotransmission in the brain and contribute to the production of the anesthetic state. In human endothelial cells, adenosine triphosphate induced a typical Ca2+change comprising of internal Ca2+release and an additional Ca2+-induced Ca2+influx from the outside.39In endothelial cells incubated with xenon, only the first part of the adenosine triphosphate–induced Ca2+response was observed, and the Ca2+-dependent Ca2+influx was absent. If xenon was removed, the cells again showed both parts of the Ca2+response.39These data indicate that xenon affects mechanisms regulating the Ca2+release–activated Ca2+channel of plasma membranes. The plasma membrane Ca2+–adenosine triphosphatase (PMCA) is one Ca2+transport system found in neurons responsible for maintaining low cytosolic calcium concentrations.40The PMCA activity is selectively inhibited by halogenated anesthetics at clinical concentrations.41In rat brain synaptic plasma membranes, xenon inhibits PMCA pump activity, resulting in an increase in neuronal Ca2+concentration and altered excitability in these cells.42In C6 rat glioma cells, PMCA activity was inhibited by xenon in clinical relevant concentrations, and this effect was potentiated in the presence of halothane.43The rate of phospholipid methylation in rat brain synaptosomal membranes is linked to the coupling of neuronal excitation to neurotransmitter release. Xenon increased phospholipid methylation and simultaneously depressed PMCA activity.44
The neurotransmitter nitric oxide may play a role in anesthetic action45; nitric oxide–dependent decrease in cyclic guanosine monophosphate occurs in halothane- and isoflurane-anesthetized rats in several brain areas. Contrastingly, xenon, like ketamine, increased cyclic guanosine monophosphate in the spinal cord, brainstem, and hippocampus,46although neuronal nitric oxide synthase activity was not altered by xenon.46Xenon exerts some effects on second messenger signaling; however, currently it is unclear how this causally relates to the production of anesthesia.
Neurotransmitter Release
The hypothalamus is a crucial homeostatic center in the brain, and the noradrenergic neuronal activity therein modulates physiologic states including consciousness and the cardiovascular system. The posterior hypothalamus is involved in the regulation of the autonomic nervous system, and an increase in norepinephrine concentration in the posterior hypothalamus increases sympathetic tone. In rats, xenon stimulates noradrenergic neurons in the hypothalamus more potently than does nitrous oxide, as measured by microdialysis in rats.47This may be one mechanism contributing to the hypnotic and the sympathotonic effects of xenon.
In the rat cerebral cortex, xenon induced an initial increase in ACh release, followed by a gradual decrease, as measured by brain microdialysis in vivo .48In addition, xenon had no effect on acetylcholinesterases measured in vitro .49Currently, the relevance of xenon’s effects on the cholinergic system to the mechanisms of anesthesia, amnesia, analgesia, and organoprotection are unknown and requires further study.
Putative Sites for Antinociception
The precise mechanism for the antinociceptive effect of xenon remains to be elucidated. In spinal cord–intact cats, xenon suppressed spinal cord dorsal horn neurons.50Xenon directly inhibited the nociceptive responsiveness of spinal dorsal horn neurons in spinally transected cats51; therefore, unlike nitrous oxide, xenon’s antinociceptive action does not require the involvement of the descending inhibitory system.52In support of this, Ohara et al. 53reported that xenon exerted potent antinociceptive action in rats independently of opioidergic or adrenergic receptors. Rather, there seems to be a direct suppression of polysynaptic transmission within the dorsal horn as reflected by a diminution in the slow ventral root response to stimulation of the primary afferents by xenon.54As a generic class, the NMDA receptor antagonists induce profound analgesia, and this may be the mechanism for xenon’s analgesic properties. Consistent with this premise, xenon has been shown to reduce formalin-induced nociception and hyperalgesia at various ages, indicating an age-independent antinociceptive profile, unlike nitrous oxide, which is ineffective in the young.6,8Xenon exerts potent antinociceptive action at the level of the spinal cord, but the above studies do not exclude contribution from supraspinal sites (in the intact animal); e.g. , activation of the midbrain reticular network with xenon may indicate activation of the supraspinal antinociceptive system.55
Neuroprotection
N -methyl-d-aspartate receptors play a pivotal role in the propagation of acute neuronal injury56; hence, many have advocated the use of NMDA antagonists to interrupt the pathogenesis of acute neuronal injury. Xenon has been shown to be surprisingly potent as a neuroprotectant in a variety of in vitro and in vivo models. Crucially, xenon provides marked protection against injury well below anesthetic concentrations, with IC50concentrations in some models as low as 10–20{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} of an atmosphere. Xenon decreases acute neuronal injury in response to both the exogenous administration of excitotoxins or through deprivation of oxygen and glucose in a neuronal–glial mouse coculture system.57,In vivo , xenon prevents the morphologic and functional consequences of acute neuronal injury provoked by ischemia (middle cerebral artery occlusion) in adult mice,58cardiopulmonary bypass in adult rats,59and excitotoxins in adult rats.57
Many NMDA receptor antagonists may reduce the neuronal damage after cerebral ischemia but concomitantly produce psychotomimetic side effects.60,61These effects were observed after ketamine and nitrous oxide administration, but not after xenon.62A reliable marker of neuronal toxicity is the c-Fos expression in distinct cerebral regions.63Xenon, in contrast to nitrous oxide or ketamine, does not induce c-Fos expression in the retrosplenial and posterior cingulate nuclei in rats in vivo .62It is also possible that the combined use of NMDA receptor antagonists may exacerbate neurotoxicity. Nagata et al. 64demonstrated that nitrous oxide alone produced a small amount of c-Fos expression but significantly enhanced ketamine-induced neurotoxicity. In contrast, xenon alone exhibited no neurotoxicity and concentration-dependently reduced the ketamine-induced c-Fos expression in rat posterior cingulate and retrosplenial cortices.64
Hypothermia is the only therapeutic intervention that has, so far, been shown to provide even a modicum of neuroprotection in the clinical setting65,66; therefore, we sought to determine the possible convergence of hypothermia and xenon on similar signaling pathways. When applied individually, both xenon and hypothermia reduced acute neuronal injury after oxygen–glucose deprivation. When applied together, the neuronal protection provided by the combination was significantly greater than could be expected from a simply additive interaction (fig. 1). Such a synergistic interaction with hypothermia may be a unique feature of xenon because it is not present with another NMDA receptor antagonist, gavestinel. A Van’t Hoff analysis revealed that a surprisingly large increase in the enthalpy is associated with hypothermia-induced reduction in injury-induced lactate dehydrogenase release when xenon is present, which is considerably larger than could plausibly be attributed to the enthalpy of binding of xenon to its putative site(s) on the NMDA receptor, and that more complex mechanisms must be operating. Using an in vivo neonatal rat model of hypoxic–ischemic injury, the synergistic interaction of the two neuroprotectant interventions was confirmed using both morphologic and functional measures of outcome.67
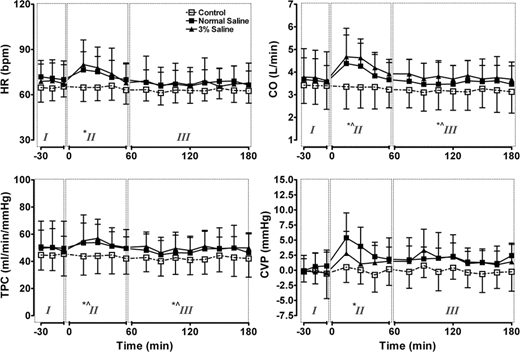
Fig. 1. Ratio of the right hemispheric weight to that of the left determined in 7-day-old postnatal rat pups after right common carotid artery ligation and exposure to 8{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} oxygen in either nitrogen or xenon for 90 min at 37°C. Whereas 2°C of hypothermia and 20{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} xenon provided no significant neuroprotection when applied separately, they provided substantial neuroprotection (less brain shrinkage) when combined. Reproduced with permission. 67
Putative Mechanisms for Xenon’s Neuroprotective Properties
Xenon exerts its neuroprotective effect through an antiapoptotic mechanism67and did not produce apoptotic neurodegeneration in neonatal rats.68Using flow cytometry of sorted cultured mouse neurons, xenon’s neuroprotective action seems also to be mediated via antiapoptotic pathways (fig. 2). Brief (10-min) exposure of cortical neuronal cells in primary culture to glutamate resulted in a significant decrease in cell viability (23 ± 8{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}) when assessed 24 h later by fluorescent-activated cell sorting after staining with propidium iodide (for cell death) and annexin V (for apoptosis). Exposure to xenon doubled the number of viable cells, and this improvement exclusively resulted from a reduction in the amount of apoptosis (fig. 3). Necrotic cell death, on the other hand, was not reduced with xenon exposure when compared with the control group. A similar antiapoptotic effect of xenon was noted when injury was acute neuronal injury provoked by NMDA exposure or by oxygen–glucose deprivation.
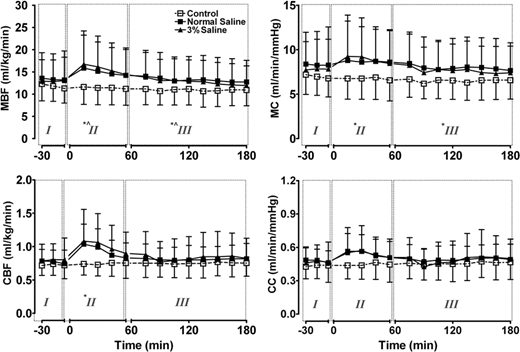
Fig. 2. Flow cytometry of sorted cultured mouse neurons stained with propidium iodide (for cell death) and annexin V (for apoptosis). ( A ) Untreated controls. Brief exposure of cortical neuronal cells in primary culture to glutamate (300 μm) resulted in a significant decrease in cell viability ( B ). Exposure to xenon doubled the number of viable cells ( C ), and this improvement was exclusively due to a reduction in the amount of apoptosis ( D ). ** P < 0.01 versus control. Reproduced with permission. 67
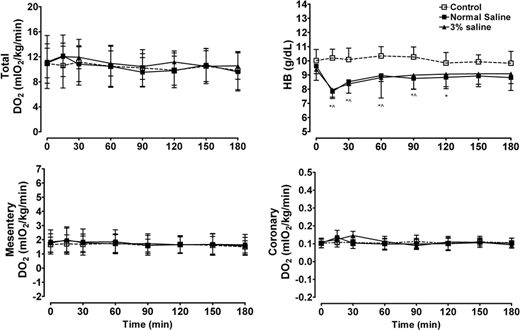
Fig. 3. Xenon attenuates apoptotic cell death in rat pups injured by hypoxia–ischemia in vivo as assessed from stained sections from both the cortex and the hippocampal gyrus 24 h after injury. Hypoxia–ischemia was induced in 7-day-old postnatal rat pups by right common carotid artery ligation and exposure to 8{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} oxygen in nitrogen. Xenon was administered for 90 min at 37°C 4 h after the injury. * P < 0.05. Reproduced with permission. 67
Xenon’s antiapoptotic effect was also confirmed in in vivo studies (fig. 3). In rat pups injured by hypoxia–ischemia, xenon alone, as well as a neuroprotective combination of subtherapeutic interventions with xenon (20{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}) and hypothermia (35°C), significantly increased cell viability by decreasing apoptosis as assessed by morphologic criteria. These data were corroborated by immunoblotting that established a decrease in the proapoptotic factor Bax and an increase in the antiapoptotic factor BclXL(fig. 4).
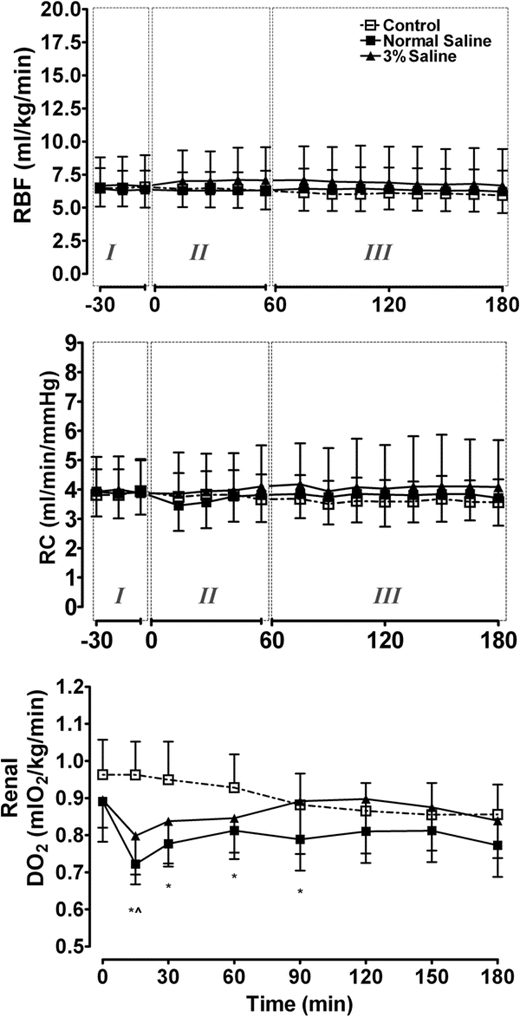
Fig. 4. Western blots showing the effects of xenon and hypothermia treatment on expression of certain proteins associated with apoptotic pathways (Bax, BclXL). Four hours after hypoxia–ischemia induced by right common carotid artery ligation and exposure to 8{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} oxygen in nitrogen, 7-day-old postnatal rat pups were treated with xenon and/or hypothermia for 90 min. After recovery for 24 h, the animals were killed and the brains were harvested. Xenon alone at 70{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} or 20{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} xenon accompanied with modest hypothermia (35°C) caused an increase in the antiapoptotic factor BclXL( A ) and a significant reduction in the proapoptotic factor Bax ( B ) in the ipsilateral cerebral hemisphere to the injury. * P < 0.05. ** P < 0.01. Reproduced with permission. 67
Xenon also interacts synergistically with isoflurane, another anesthetic capable of providing neuroprotection. Neuroprotection of isoflurane is at least in part a result of GABAAreceptor stimulation,69and the potentiated neuroprotective effect of a combination with xenon may be due to their differing mechanisms of action. Consistent with this concept, NMDA-induced Ca2+influx, which is thought to be a critical event involved in excitotoxic neuronal death,70was reduced after administration of xenon in cortical cell cultures.71
The striatum is a subcortical structure mostly resistant to neuroprotective interventions. Xenon at 50{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}, but not nitrous oxide, reduced ischemic brain damage in the striatum. However, David et al. 71also showed an intriguing effect at higher concentrations of xenon (75{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5}); xenon at this concentration was not neuroprotective. Despite the reported “potentially neurotoxic” effect of xenon, no evidence was presented to support this statement; no difference in infarct volume was observed between 75{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} xenon and controls.
In addition to the effects mediated via the NMDA receptor, xenon protects cortical neurons against hypoxia-related cell damage via Ca2+-dependent mechanisms.72Petzelt et al. 73demonstrated in dopaminergic neurons a xenon-induced neuroprotection. Nerve growth factor–differentiated pheochromocytoma cells (PC-12 cells) include D1– and D2-dopamine receptors and release dopamine as a result of increased release and reduced uptake rate of dopamine after hypoxia. This dopamine release is linked to cellular damage as evidenced by lactate dehydrogenase release from the cells. Xenon prevented the dopamine release in PC-12 cells induced by 2 h of hypoxia, and this neuroprotective effect was reduced after buffering intracellular Ca2+using a Ca2+chelator.73This is of special interest because NMDA antagonist neurotoxicity has been linked to excess dopaminergic activation,62and xenon, which itself lacks toxicity62and protects against ketamine induced neurotoxicity,64seems to prevent dopamine induced toxicity. The role of dopamine in the mechanism of NMDA antagonist toxicity and xenon’s neuroprotective effects requires further investigation.
In a neuronal–glial cell coculture, preexposure to xenon for 2 h caused a concentration-dependent reduction of lactate dehydrogenase release from cells deprived of oxygen and glucose 24 h later; xenon’s preconditioning effect was abolished by cycloheximide, a protein synthesis inhibitor.74Preconditioning with xenon decreased propidium iodide staining in a hippocampal slice culture model subjected to oxygen–glucose deprivation. In an in vivo model of neonatal asphyxia involving hypoxic–ischemic injury to 7-day-old rats, preconditioning with xenon reduced infarction size when assessed 7 days after injury, and sustained improvement in neurologic function was still evident after 30 days. Contrastingly, we observed no preconditioning with nitrous oxide.74From our in vivo experiments, quantitative immunoblotting revealed that the phosphorylated Ca2+/cAMP-responsive element binding protein and brain-derived neurotrophic factor74are significantly up-regulated after xenon exposure, with a time course similar to that of the preconditioning response; this provides an important clue as to which signaling pathways are involved. Neither brain-derived neurotrophic factor nor the phosphorylated Ca2+/cAMP-responsive element binding protein levels changed after nitrous oxide exposure.74The molecular effects of xenon on the central nervous system are summarized in table 1.

Putative Sites for Xenon on the Cardiovascular System
In isolated guinea pig hearts, 40–80{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} xenon did not significantly alter heart rate, atrioventricular conduction time, left ventricular pressure, coronary flow, oxygen extraction or consumption, cardiac efficiency, or flow responses to bradykinin.75In isolated cardiomyocytes, the amplitudes of the Na+, the L-type Ca2+, and the inward-rectifier K+channel were not altered by 80{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} xenon, suggesting that it does not affect the cardiac action potential.75These results indicate that xenon has no physiologically important effects on the guinea pig heart.
Electrophysiologic studies on cardiomyocytes revealed that halogenated anesthetics depress Ca2+currents through L-type Ca2+channels, thereby producing negative inotropic effects and shorten the duration of the action potential.76In human atrial myocytes, xenon at a concentration of 70{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} did not depress L-type Ca2+currents, as measured by patch clamp techniques.77Voltage-gated potassium currents are responsible for the repolarization of cardiomyocytes and influence the timing of the refractory period. Transient potassium outward currents were only slightly inhibited, and sustained potassium currents were not affected by xenon.77
In vivo , xenon had minor direct negative inotropic effects when administered selectively into the coronary artery system using a coronary bypass system.10,In vitro , xenon neither depressed myocardial contractility nor influenced the positive inotropic stimulation of isoproterenol or the force-frequency relation in cardiac muscle bundles78; these effects are consistent with xenon’s stable cardiovascularly profile.5
Cardioprotection
Xenon also has cardioprotective effects: Given during reperfusion, it reduced infarct size after regional myocardial ischemia in rabbits in vivo .79Application of a substance after ischemia during initial reperfusion was recently termed “postconditioning.” Xenon can also induce cardioprotection via the “preconditioning” mechanism (whereby a previous stimulus or stressor provides protection against a later injury). Ischemic preconditioning describes the protection of myocardial tissue against infarction by short, nonlethal periods of ischemia. In the past years, the halogenated (volatile) anesthetics, e.g. , isoflurane80,81or sevoflurane,82have been recognized to mimic the strong cardioprotection exerted by ischemic preconditioning (pharmacologic or anesthetic-induced preconditioning). Pharmacologic activation of different receptors mimics ischemic preconditioning and activates inhibitory G proteins83and protein kinase C (PKC) (for details, see fig. 5).84This activation of PKC affects other signaling pathways, such as Raf-MEK1-MAP kinases and the PI3-kinase-Akt cascade.85Moreover, the release of free radicals activates different kinases, including PKC (mainly its ϵ-isoform),86tyrosine kinases,87and mitogen-activated protein kinases (MAPKs),88which act as triggers and/or mediators of the resulting cardioprotection (for review, see Das et al. 89). Recent data indicate that also xenon is able to induce preconditioning of the heart in vivo . In anesthetized rats subjected to 25 min of coronary artery occlusion followed by 120 min of reperfusion, either xenon or isoflurane was administered during three 5-min periods before ischemia.9Xenon inhalation resulted in a significant reduction of the infarct size compared with controls. Calphostin C, an inhibitor of PKC, and the p38 MAPK inhibitor SB203580 abolished the preconditioning effects of xenon and isoflurane. These data suggest that PKC and p38 MAPK are key mediators of xenon-induced preconditioning. PKC-ϵ is one of the isoforms present in cardiac myocytes and is mainly implicated in preconditioning mechanisms. PKC isoforms have been shown to be mainly regulated via translocation to different cell compartments and subsequent phosphorylation, resulting in their activation. By use of a phosphospecific antibody against PKC-ϵ, it was demonstrated that xenon leads to a marked phosphorylation of PKC-ϵ compared with controls.9Calphostin C abolished the effect of xenon on PKC-ϵ phosphorylation. PKC-ϵ translocates from cytosolic to membrane regions upon different stimuli. Both xenon and isoflurane increased the amount of PKC-ϵ in the membrane fraction compared with controls. The translocation to membrane fraction could be blocked by calphostin C. By using immunohistochemical techniques, Uecker et al. 90observed that isoflurane-induced preconditioning leads to translocation of PKC-δ and PKC-ϵ to nuclei (PKC-δ and PKC-ϵ), to mitochondria (PKC-δ), and to the sarcolemma and intercalated disks (PKC-ϵ). Only phosphorylation of PKC-δ on serine643 was increased after isoflurane administration but not phosphorylation of PKC-ϵ. The PKC blockers chelerythrine and rottlerin blocked PKC activation and anesthetic-induced cardioprotection. We examined whether other than the ϵ isoforms of PKC are involved in xenon induced preconditioning.91In rat hearts in vivo , application of rottlerin, an inhibitor of PKC-δ, had no effect on infarct size. Activation of PKC isoforms during the preconditioning stimulus may be time dependent.92However, Western blot analysis showed no influence of xenon preconditioning on phosphorylation of PKC-α at four different time points during the preconditioning protocol, suggesting an isoform specific activation of PKC-ϵ by xenon.
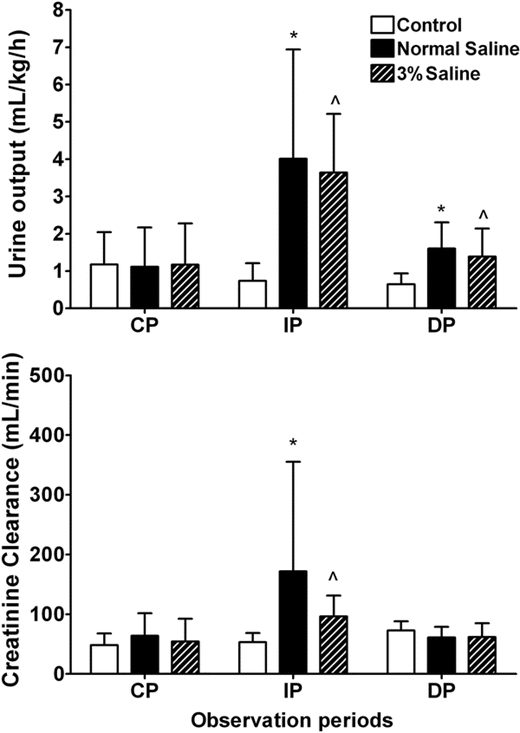
Fig. 5. Preconditioning by xenon involves the activation of protein kinase C (PKC). The effect was shown by the use of the specific PKC inhibitors staurosporine and calphostin C. Tyrosine kinases (TK) may also be mediators of cardioprotection by halogenated anesthetics, but their relation to PKC is not yet defined. In addition, the mitogen-activated protein kinase (MAPK) family (p38, JNK and ERK) seems to be involved because the blockade by the specific inhibitors PD98059 (ERK-1/2) and SB203580 (p38 MAPK) completely abolished the cardioprotection elicited by xenon. Downstream of p38 MAPK, the phosphorylation of a member of the heat shock protein (HSP) family, HSP27, is up-regulated, resulting in cytoskeleton changes in the myocytes. Whether the upstream kinases of MAPK, the MAPK kinases (MKKs) and MKK kinases (MKKKs), are involved is poorly investigated. The upstream signaling of PKC is not yet clearly defined. It remains to be determined in detail whether the activation occurs via the phospholipase C (PLC)/3-phosphoinositide–dependent kinase 1 (PDK-1) pathway involving activation of G protein–linked receptors or via opening of mitochondrial KATP(mKATP) channels and release of reactive oxygen species (ROS), or in parallel. The role of mKATPhas been extensively studied by the use of 5-hydroxydecanoate (5-HD), a specific blocker of the mKATPchannels. Alternatively, it is suggested that the activation of endothelial nitric oxide (NO) synthase (eNOS)/AKT/HSP90 complex may lead to NO release and that this in turn activates KATPchannels. AKT (PKB) = protein kinase B; ERK-1/2 = extracellular signaling regulated kinase 1 and 2; JNK = c-jun NH2-terminal kinase; l-NAME = N -nitro-l-arginine methyl ester; p38 = mitogen-activated protein kinase p38; PD98059 = blocker of ERK-1/2; PDK = phosphatidylinositol trisphosphate–dependent kinase; PLC = protein lipase C; SP600125 = blocker of JNK.
Activation of PKC affects other downstream signaling pathways like the MAPK cascade, and in this context, it has been shown that PKC-ϵ interacts with MAPK during cardioprotection. Xenon induced a significant increase of p38 MAPK phosphorylation and calphostin C abrogated this effect, demonstrating that p38 MAPK is located downstream of PKC in the signaling cascade of xenon-induced preconditioning.9p38 MAPK is suggested to interact with the actin cytoskeleton via the MAPK-activated protein kinase-2 (MAPKAPK-2) and heat shock protein (HSP) 27. Xenon preconditioning induced phosphorylation of MAPKAPK-2 and HSP27, and both effects could be blocked by calphostin C and SB203580. Xenon enhanced the translocation of HSP27 to the particulate fraction and increased F-actin polymerization. F-actin and HSP27 were colocalized after xenon preconditioning.93These data show that xenon induces cardioprotection by preconditioning and that activation of PKC-ϵ and its downstream target p38 MAPK are central molecular mechanisms involved. Xenon activates MAPKAPK-2 and HSP-27 downstream of PKC and p38 MAPK, and these data link preconditioning by xenon in the myocardium to the actin cytoskeleton.
We also investigated the role of the p44/42 MAPK (extracellular signal–regulated kinase, ERK) and the stress-activated p54/46 MAPK (SAPK/JNK) in xenon induced preconditioning.94Both kinases play a key role in differentiation and cell survival as well as in apoptosis regulation. The ERK inhibitor PD 98059 completely abolished the observed cardioprotection offered by xenon, demonstrating an involvement of ERK 1/2 in the signal transduction. Interestingly, SP 600125, a JNK inhibitor, had no effect on infarct size reduction by xenon. In addition, the phosphorylation state of SAPK/JNK was not influenced by xenon as demonstrated by Western blot analysis. These data suggest that besides the p38 MAPK, also ERK is involved in xenon preconditioning. However, the third member of the MAPK family, the SAPK/JNK, is not a mediator of xenon preconditioning, suggesting a highly specific regulation of different kinases by xenon in the myocardium.
Several investigators have demonstrated the existence of a second episode of myocardial protection (late preconditioning), which begins 12–24 h after the preconditioning stimulus and lasts for 48–72 h. In contrast to early preconditioning, the phenomenon of late preconditioning was long thought not to be induced by halogenated anesthetics.95Interestingly, there exists increasing evidence from different in vivo models that isoflurane, sevoflurane, and desflurane produce a second window of cardioprotection.96–98Preliminary results from our laboratory show that xenon also induces late cardioprotection similar to ischemic late preconditioning. However, the molecular mechanisms behind this xenon-induced late cardioprotection remain unknown and need further investigations. Molecular effects of xenon on the heart are summarized in table 2.
It would be interesting to define the most effective strategy for organoprotection. Ischemic preconditioning can reduce infarct size to less than 20{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} of the area at risk,99and anesthetic induced preconditioning reduced infarct size to approximately 30{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} of the area at risk.81Preconditioning by sevoflurane further reduced infarct size after ischemic late preconditioning.82Because of differences in species, tissue preparations, and preconditioning protocols, it is difficult to directly compare the different experimental studies with the regard to the extent of cardioprotection. In studies from our laboratory, the cardioprotection by xenon was to the same extent as the cardioprotection by isoflurane.9,93
Other Molecular Effects Exerted by Xenon
In embryonic rat brain astroglial cells, xenon produced a block of the cell cycle at metaphase, and this effect was completely reversible by slightly increasing intracellular Ca2+concentration.100In human endothelial cells, the block in the cell cycle was at the G2–M transition and at metaphase, again reversible by increasing intracellular Ca2+concentration.101Therefore, xenon interferes with Ca2+-dependent regulatory systems, but so far, no specific event or defined regulatory complex of the Ca2+signaling system has been identified.
In human whole blood in vitro , xenon did not affect the unstimulated or agonist-induced platelet glycoprotein expression, the activation of the glycoprotein IIb/IIIa receptor, or the platelet-related hemostasis, suggesting no altered platelet function.102An investigation on neutrophil and monocyte function demonstrated no effect on the respiratory burst activity of these cells but an increased phagocytosis activity of neutrophils.103Therefore, xenon preserves neutrophil and monocyte antibacterial capacity in vitro . Selectins are involved in the initial contact between neutrophils and endothelial cells. Xenon increased the removal of selectins from neutrophil surface, thereby probably inhibiting the adhesion of neutrophils to the endothelium.104This might have implications in the recruitment of neutrophils to an inflammatory site. In addition, adhesion molecule receptors are involved in the pathophysiology of ischemia–reperfusion injury. Xenon administration only during reperfusion reduced myocardial infarct size after regional ischemia in rabbits,79and modulation of neutrophil function may be one underlying mechanism. Adhesion molecules facilitate leukocyte migration into injured tissue. However, expression of adhesion molecules on mice brain endothelial cells was not affected by 75{e43154ce913794517af217fcab284b44cfe906ba9a1c4ba2ecde5a9be0395ec5} xenon, suggesting no antiinflammatory actions at the vascular endothelium.105In an isolated cardiopulmonary bypass system, xenon had no immunomodulatory effects and did not change interleukin-8 or interleukin-10 levels.106In human monocytes in vitro , xenon increased the lipopolysaccharide-induced production of tumor necrosis factor α and interleukin-6 and activated nuclear transcription factor κB.107In contrast, isoflurane inhibited activation of nuclear transcription factor κB.
Conclusion
Xenon exerts several interesting properties including pronounced organoprotective effects in various experimental settings against ischemia–reperfusion injury of the heart and the brain. Whether this translates to a clinical benefit must be rapidly determined because preservation of myocardial and cerebral function may outweigh the significant cost of xenon administration. Clinical trials to assess the impact of xenon in settings with predictable injury such as cardiopulmonary bypass and neonatal asphyxia should be designed and underpinned with investigation of the molecular targets involved in the mechanism of action of xenon-induced neuroprotection and cardioprotection.
